【东大新闻网11月1日电】(通讯员 崔煜)近日,东南大学物理学院王金兰教授团队在单原子催化剂结构-稳定性关系研究中取得新进展,研究成果以“Structure-Stability Relation of Single-Atom Catalysts under Operating Conditions of CO2 Reduction(CO2还原工况条件下单原子催化剂的结构-稳定性关系)”为题发表在化学类顶级期刊《美国化学会会志》(Journal of the American Chemical Society)上。
单原子催化剂以其超高的原子利用率和优异的催化活性备受瞩目。但在反应条件下,单原子催化剂中的金属原子易浸出、聚集,从而显著降低催化剂的活性和选择性。因此,单原子催化剂的稳定性问题,成为制约其工业化应用的关键因素。材料的“构-效”关系能够关联材料的微观结构与宏观性能,因此,构建单原子催化剂的结构-稳定性关系,对其稳定性提升至关重要。但催化剂稳定性评估的难度远超活性和选择性,导致结构-稳定性关系的研究极具挑战。
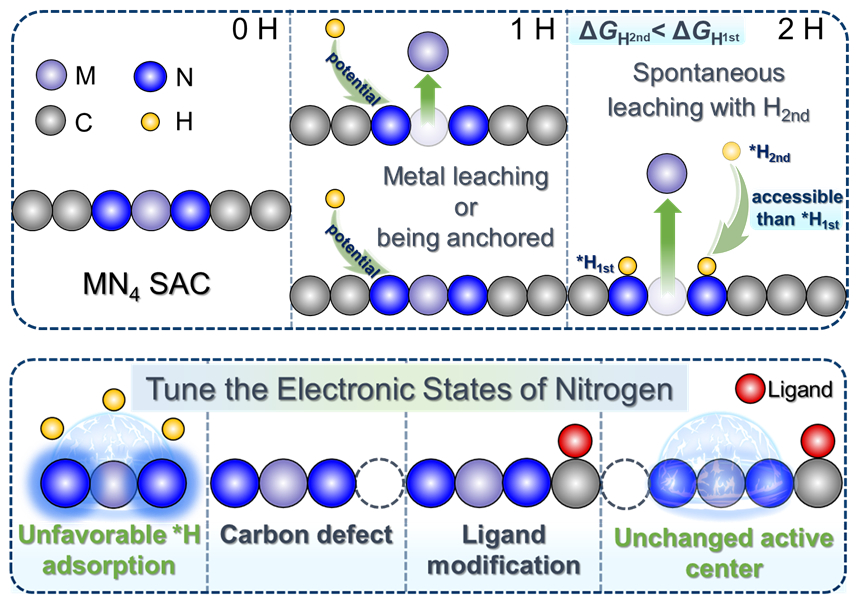
针对这一问题,团队通过恒电位密度泛函理论模拟,构建了CO2还原工况条件下单原子催化剂的结构-稳定性关系。团队采用机制引导策略,在前期提出的单原子催化剂金属浸出机制(J. Am. Chem. Soc., 2022, 144, 17140)基础上,进一步发现配位N原子上第一个氢原子的吸附与否是金属原子浸出的决定因素。基于此,团队开发出一个新的描述符,涵盖元素特性和反应条件,能够有效预测电催化环境下单原子催化剂的稳定性及金属浸出电位。进一步,团队提出数种在不改变活性中心几何结构的前提下增强单原子催化剂稳定性的策略,并获得实验数据支持。以上研究结果为单原子催化剂稳定性的快速评估和针对性优化提供了有力工具。
本文的第一作者是东南大学博士生崔煜,东南大学物理学院王金兰教授及凌崇益副教授为通讯作者。该工作受到国家重点研发计划、国家自然科学基金及江苏省基金等项目的资助。
论文链接:https://doi.org/10.1021/jacs.4c11516
供稿:物理学院
(责任编辑:刘明源 审核:宋健刚)
